
研究
About basic research
基礎研究について
循環器基礎研究
循環器内科では実臨床に基づく基礎研究、トランスレーショナルリサーチを行っています。これは臨床現場で直面した疑問点・問題点をもとに基礎研究を計画・立案し、そして基礎研究で得られたデータ・成果を最終的には臨床に還元し、医学・医療の進歩に資することを目的としています。
ここでは当科で取り組んでいる代表的な基礎研究を御紹介します。
ここでは当科で取り組んでいる代表的な基礎研究を御紹介します。
心腎連関
生理的状態において心機能と腎機能は互恵関係にあり、全身の恒常性維持に大きく貢献しています。しかし一度いずれかの臓器障害が発症すると、互いに増悪因子として悪循環を形成してしまいます。我々は心血管疾患(CVD:
cardiovascular disease)と慢性腎臓病(CKD: chronic kidney
disease)の関係に代表されるこの「心腎連関」あるいは「心腎症候群」と呼ばれる病態に着目し、その分子機序や病態の解明とともに、効果的な治療法の提案も含め研究を進めてきました。
我々は慢性炎症を惹起し動脈硬化を促進するPlacental growth factor (PLGF)、およびPLGFの内因性阻害因子として働く可溶型Fms様チロシンキナーゼ1 (sFlt-1)に着目し研究を進めて来ました。このsFlt-1産生が腎機能障害とともに低下し、結果としてCKDの際に生じるPLGF/sFlt-1比の上昇が慢性炎症を介してCVDを増悪させることを臨床検体および基礎実験を通じて証明し、sFlt-1の補充、PLGFあるいはPLGF/sFlt-1比上昇時に活性化するMCP-1の抑制、尿毒症物質低下効果のある経口吸着炭 (AST-120、クレメジン)投与による治療の可能性や、圧負荷時にのみsFlt-1ノックアウトマウス特異的に低下するLong non-coding RNA が存在することを見出し、発表してきました(図1)。
我々は慢性炎症を惹起し動脈硬化を促進するPlacental growth factor (PLGF)、およびPLGFの内因性阻害因子として働く可溶型Fms様チロシンキナーゼ1 (sFlt-1)に着目し研究を進めて来ました。このsFlt-1産生が腎機能障害とともに低下し、結果としてCKDの際に生じるPLGF/sFlt-1比の上昇が慢性炎症を介してCVDを増悪させることを臨床検体および基礎実験を通じて証明し、sFlt-1の補充、PLGFあるいはPLGF/sFlt-1比上昇時に活性化するMCP-1の抑制、尿毒症物質低下効果のある経口吸着炭 (AST-120、クレメジン)投与による治療の可能性や、圧負荷時にのみsFlt-1ノックアウトマウス特異的に低下するLong non-coding RNA が存在することを見出し、発表してきました(図1)。
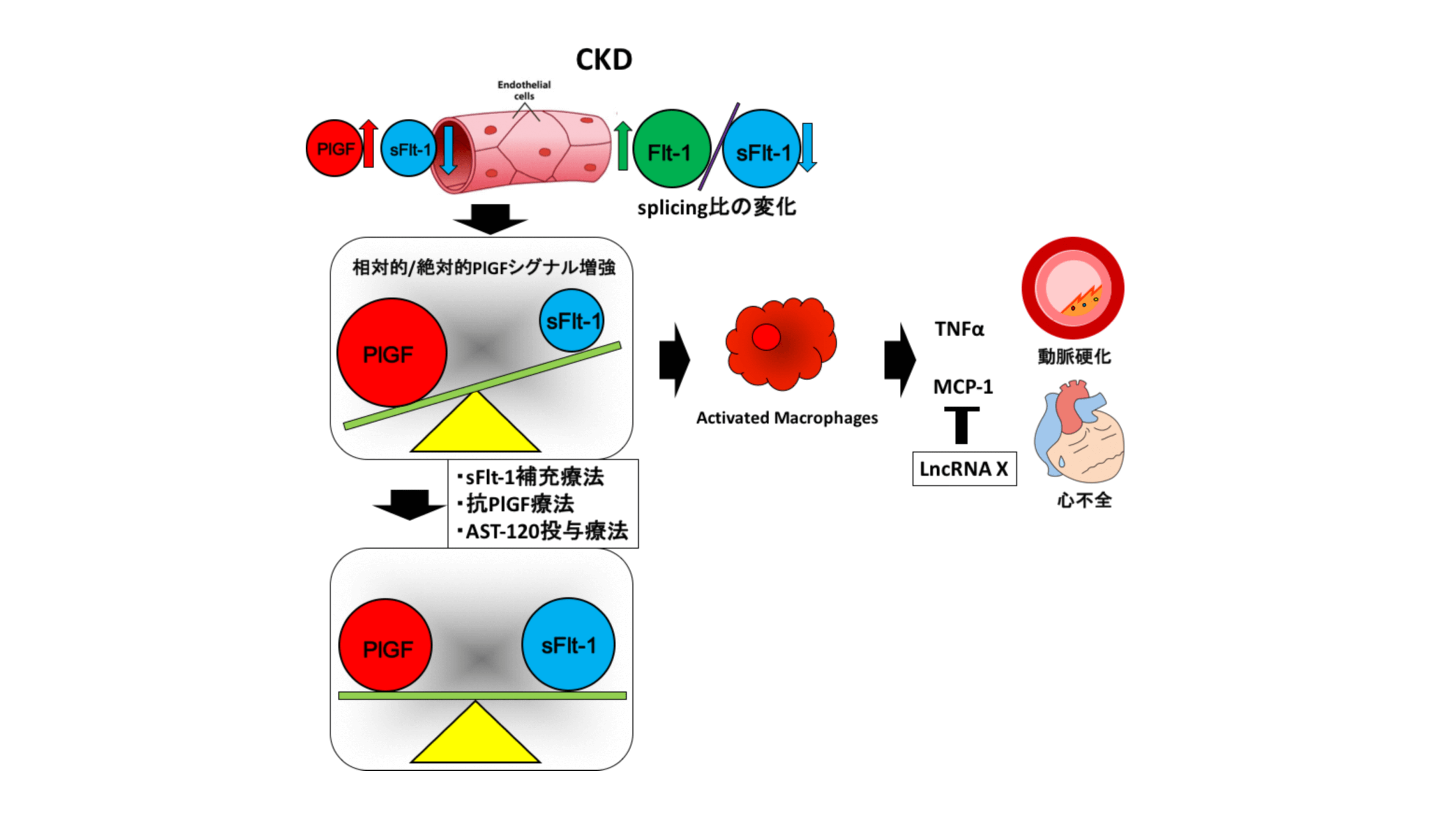
図1 心腎連関分子メカニズムにおけるsFlt1, PLGFの役割と介入ポイント
心筋疾患・心不全
心筋疾患を病理所見から始まり、その分子メカニズムやゲノム情報に至るまで幅広く解析を行っています。
心筋症に関する研究としては、重症の拡張型心筋症を生じるラミン遺伝子(LMNA)異常による拡張型心筋症発症メカニズムの解明に取り組むとともに、心筋症モデルマウスを用いた新規治療法開発に取り組んでいます。Lmnaノックアウトマウスを用いた拡張型心筋症の発症機序の解明(図2)、遺伝子異常により不足するラミン蛋白をアデノ随伴ウイルス(AAV; Adeno-associated virus)を用いて発現させる新規治療法などを検討し発表を行ってきました。
心筋症に関する研究としては、重症の拡張型心筋症を生じるラミン遺伝子(LMNA)異常による拡張型心筋症発症メカニズムの解明に取り組むとともに、心筋症モデルマウスを用いた新規治療法開発に取り組んでいます。Lmnaノックアウトマウスを用いた拡張型心筋症の発症機序の解明(図2)、遺伝子異常により不足するラミン蛋白をアデノ随伴ウイルス(AAV; Adeno-associated virus)を用いて発現させる新規治療法などを検討し発表を行ってきました。
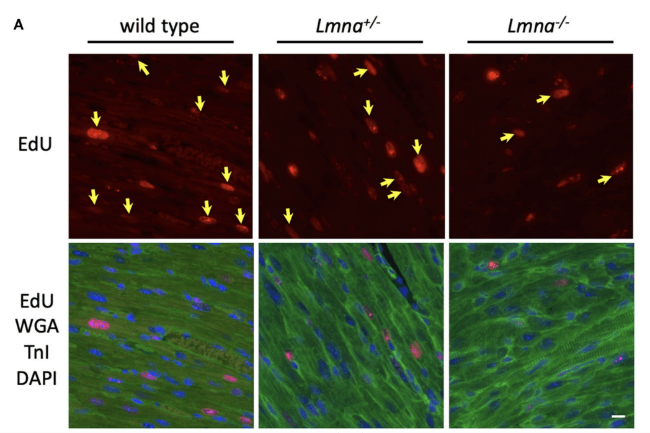
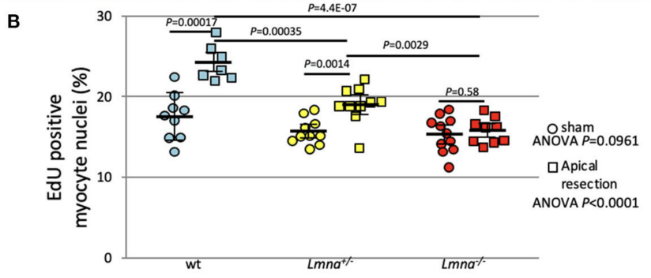
図2 Lmna ノックアウトマウスにおける心筋障害後心筋細胞増殖能
生後2日目で心尖部を切除するとwild typeマウスで生じる心筋細胞増殖能(EdU取り込みで評価)は、Lmnaヘテロノックアウトマウスで減弱し、Lmnaホモノックアウトマウスではほどんど認められない。このような心筋障害後再生能力の差が蓄積し拡張型心筋症発症に結びつくと考えられた。
心不全に関しては、カテコラミン負荷による急性心不全(たこつぼ症候群モデル、およびClinical Scenario
1モデル)について臨床検体および動物実験による検討を行って来ました。たこつぼ症候群は、一過性の心収縮能低下を特徴とする疾患群で、身体的・精神的ストレスを背景として急性冠症候群に類似した胸痛などの症状で突然発症し、数週間の経過でほぼ正常化するという経過をたどる基本的には予後良好な疾患ですが、重症急性心不全を呈する症例や、心破裂から死亡に至る症例も存在します。その発症機序については多枝冠動脈攣縮説、急性冠微小循環障害説などが考えられていますが、カテコラミン毒性による心臓交感神経系の過活動およびそれに引き続く心筋収縮不全が関与しているとする説が有力視されています。我々は当院で解析したたこつぼ症候群心筋生検組織結果から、その発症機序にβ受容体脱感作の関与があることを世界で初めて組織学的に証明することに成功しました(図3)。
また、Clinical Scenario 1に代表される急性心不全の発症機序に、血管平滑筋におけるβ受容体脱感作が関与していることを、たこつぼ症候群発症にも関与する蛋白に着目し証明しました。GRK2を血管平滑筋特異的に過剰発現させたマウスを作成し、カテコラミン負荷によりClinical Scenario 1に類似した後負荷上昇による急性心不全モデルマウス作成に成功し、同症発症メカニズムの基礎的検討を進め、発表を行っています(図4)。
また、Clinical Scenario 1に代表される急性心不全の発症機序に、血管平滑筋におけるβ受容体脱感作が関与していることを、たこつぼ症候群発症にも関与する蛋白に着目し証明しました。GRK2を血管平滑筋特異的に過剰発現させたマウスを作成し、カテコラミン負荷によりClinical Scenario 1に類似した後負荷上昇による急性心不全モデルマウス作成に成功し、同症発症メカニズムの基礎的検討を進め、発表を行っています(図4)。
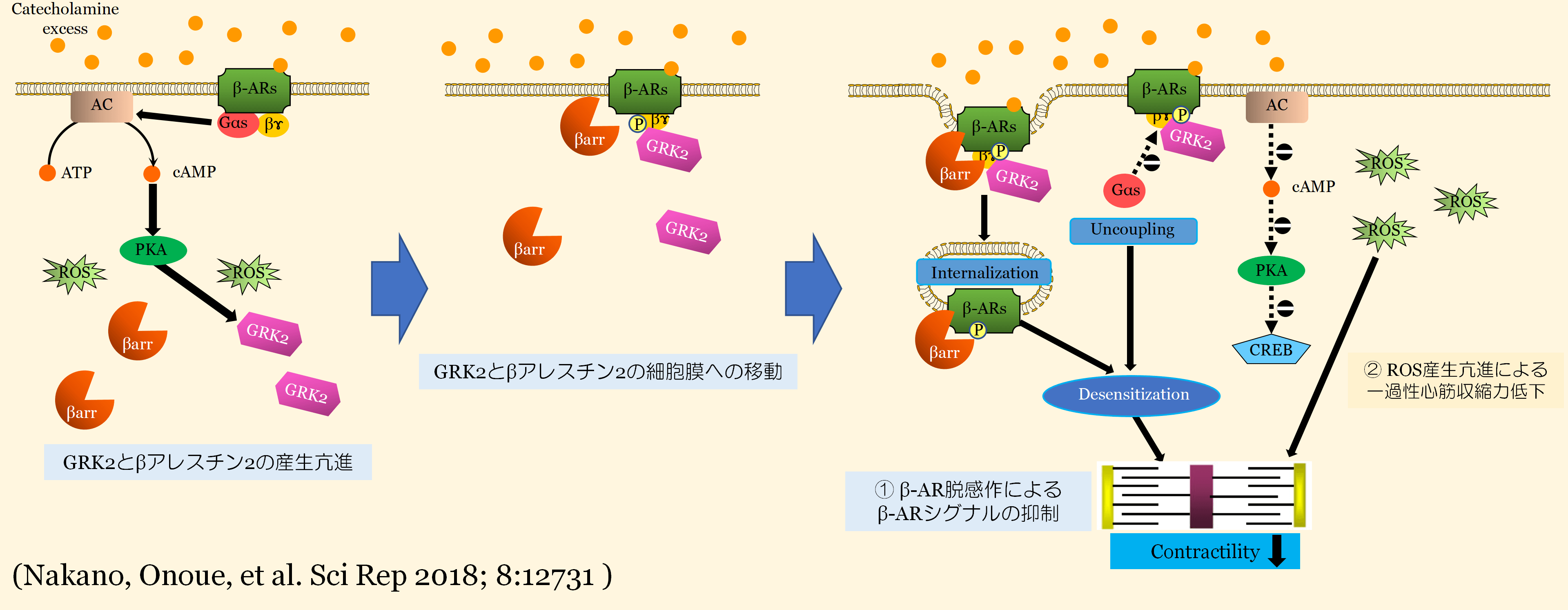
図3 たこつぼ心筋症発症機序概念図
過剰なカテコラミン刺激により心筋細胞β受容体は一過性に脱感作を生じ心筋収縮は抑制される。
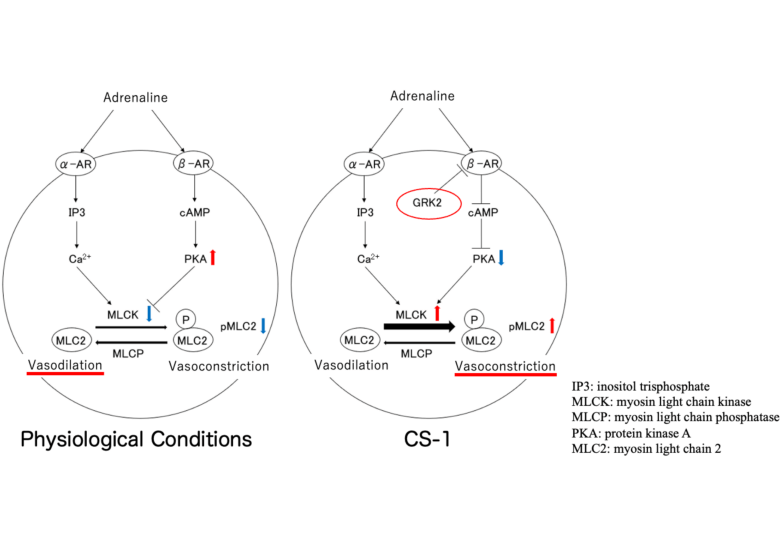
図4 Clinical Scenario 1急性心不全発症機序概念図
GRK2が過剰に存在すると血管平滑筋β受容体は脱感作を生じ、カテコラミン刺激により生理的状況とは異なり、強い血管収縮に向かう。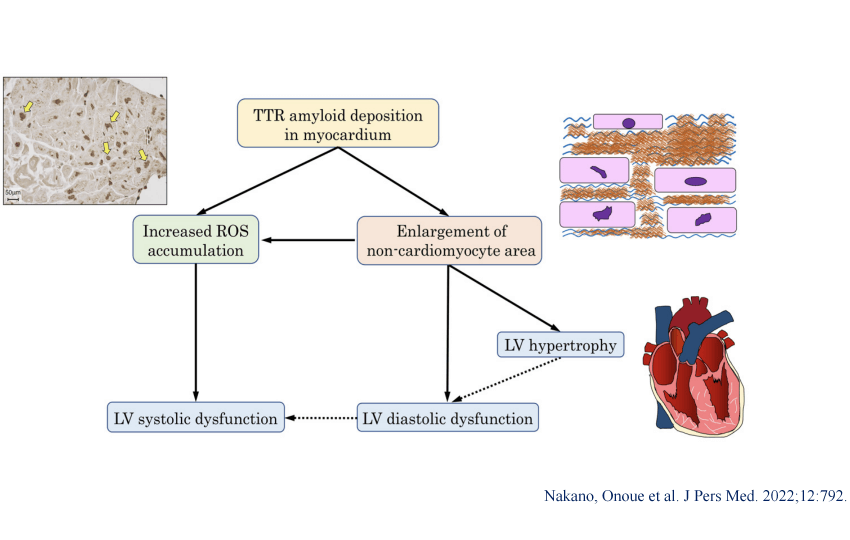
図5 ATTR心アミロイドーシスの進展機序
アミロイド線維の沈着により心筋間質の線維化が促進され、心筋細胞の活性酸素種が増加し、心機能低下を生じる。劇症型心筋炎
急性心筋炎の中でも、血行動態の悪化により昇圧剤の投与や機械的循環補助を必要とする重症病態である、劇症型心筋炎について、全国255施設にご協力頂き2021年1月末までに819症例という世界最大レジストリを構築し検討を行っています。このレジストリの臨床背景をまとめるとともに、心筋生検組織の実際のスライドを289例借り受け、その組織学的特徴を発表し重症化と関連する因子などの詳細な検討を進めています。
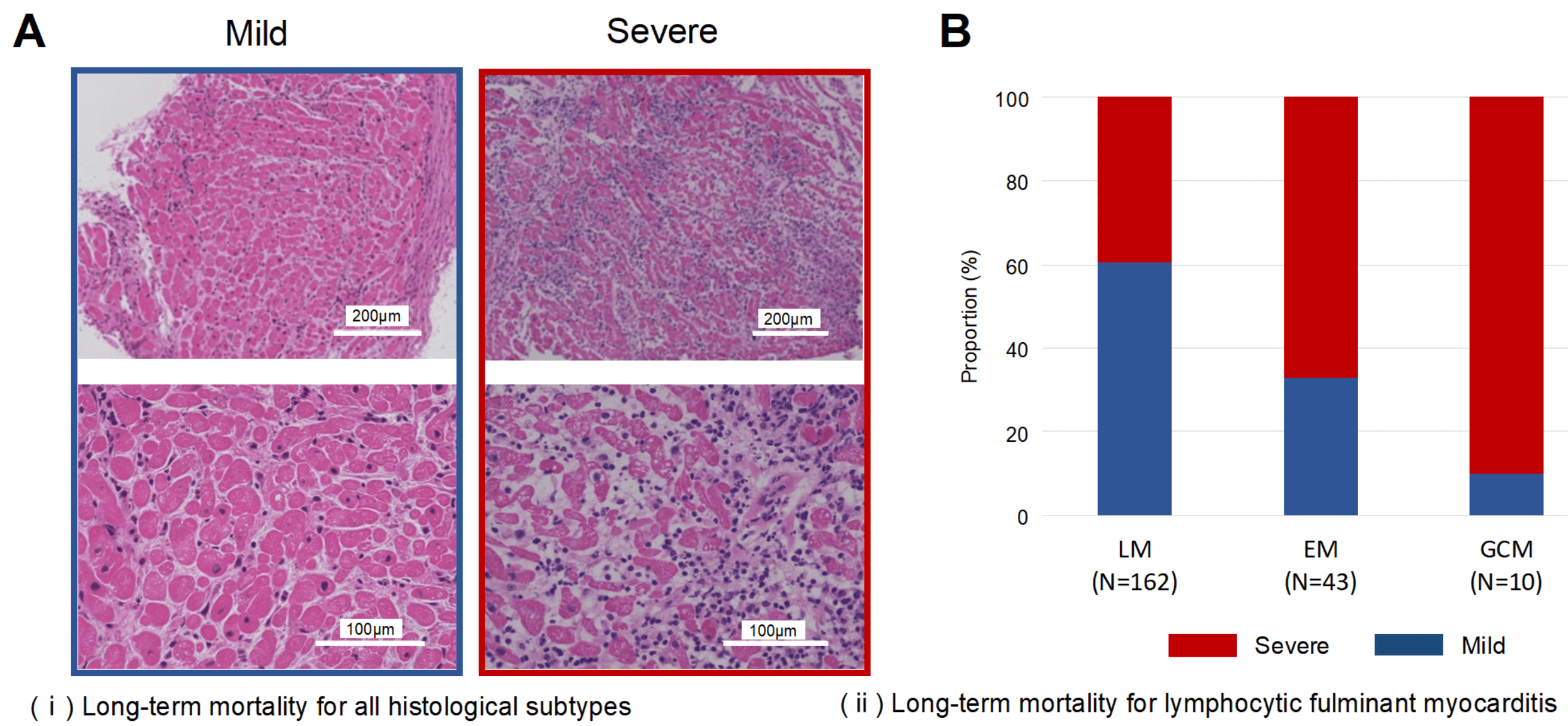
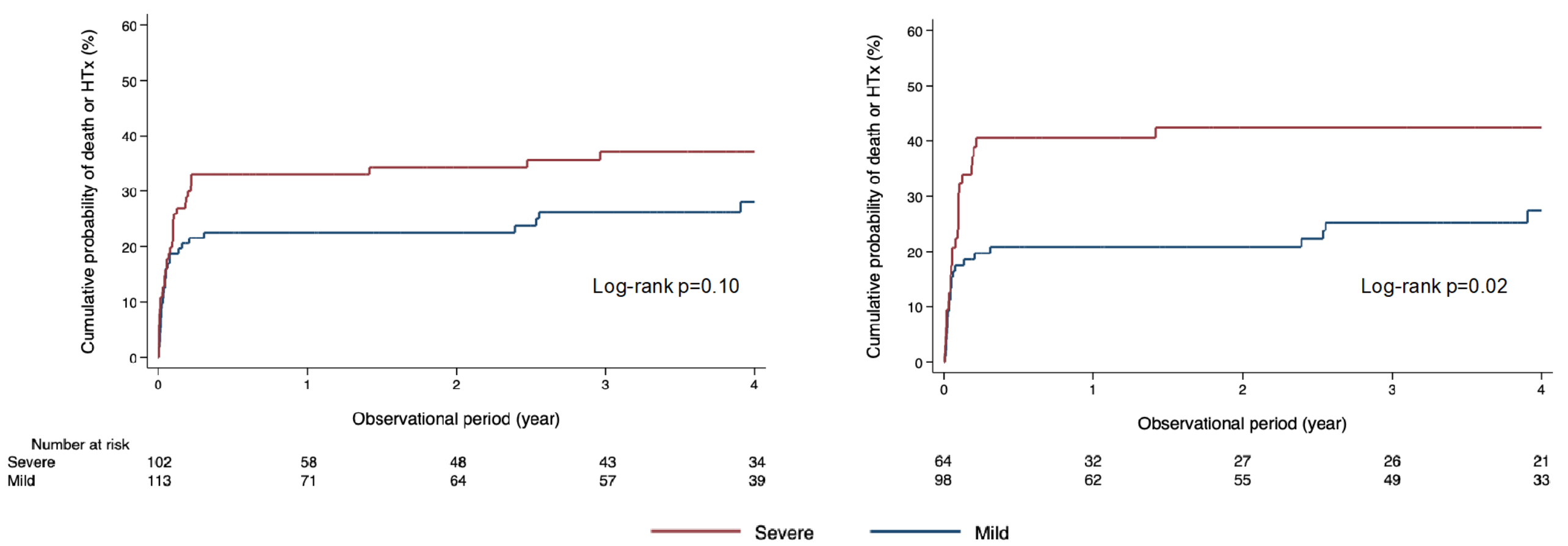
図6 劇症型心筋炎の組織学的検討
リンパ急性心筋炎(LM)、好酸球性心筋炎(EM)、巨細胞性心筋炎(GCM)各病型毎の重症度と予後。特にLMでは心筋細胞変性が高度なほど予後が悪いことがわかった。神経体液性因子
ナトリウム利尿ペプチドシグナルを中心とした研究で、ナトリウム利尿ペプチドとミネラルコルチコイドレセプター(MR)の相互作用、心不全の治療薬として注目を集めているARNIに関連した研究を進めています。
ナトリウム利尿ペプチドを分解するネプリライシン(NEP)の阻害により、ナトリウム利尿ペプチドの作用は増強します。数倍に増加した循環血液中のANP・BNPが血管拡張や利尿などに作用していると考えられており、もちろんその全身的効果は重要であると思われます。しかし、これまでの当科の研究ではANP・BNPが直接的に心臓局所に作用して心臓リモデリングを改善することを報告しており、NEP阻害薬のターゲット臓器の1つは心臓であると考えていました。我々は全身のNEP受容体欠損マウスと,心筋細胞でのNEP過剰発現マウスを作製し,大動脈を縮窄して圧負荷モデルを作製し心臓に及ぼす影響を検討してきました。その結果、血中より100-1000倍濃いとされる心臓組織のANP・BNPに対してNEPが及ぼす作用を確かめ、心臓リモデリングにおいては心臓局所でのNEPの作用が重要であることを確認し報告しました(図8)。さらに臨床効果の機序解明に鋭意取り組んでいます。
ナトリウム利尿ペプチドを分解するネプリライシン(NEP)の阻害により、ナトリウム利尿ペプチドの作用は増強します。数倍に増加した循環血液中のANP・BNPが血管拡張や利尿などに作用していると考えられており、もちろんその全身的効果は重要であると思われます。しかし、これまでの当科の研究ではANP・BNPが直接的に心臓局所に作用して心臓リモデリングを改善することを報告しており、NEP阻害薬のターゲット臓器の1つは心臓であると考えていました。我々は全身のNEP受容体欠損マウスと,心筋細胞でのNEP過剰発現マウスを作製し,大動脈を縮窄して圧負荷モデルを作製し心臓に及ぼす影響を検討してきました。その結果、血中より100-1000倍濃いとされる心臓組織のANP・BNPに対してNEPが及ぼす作用を確かめ、心臓リモデリングにおいては心臓局所でのNEPの作用が重要であることを確認し報告しました(図8)。さらに臨床効果の機序解明に鋭意取り組んでいます。
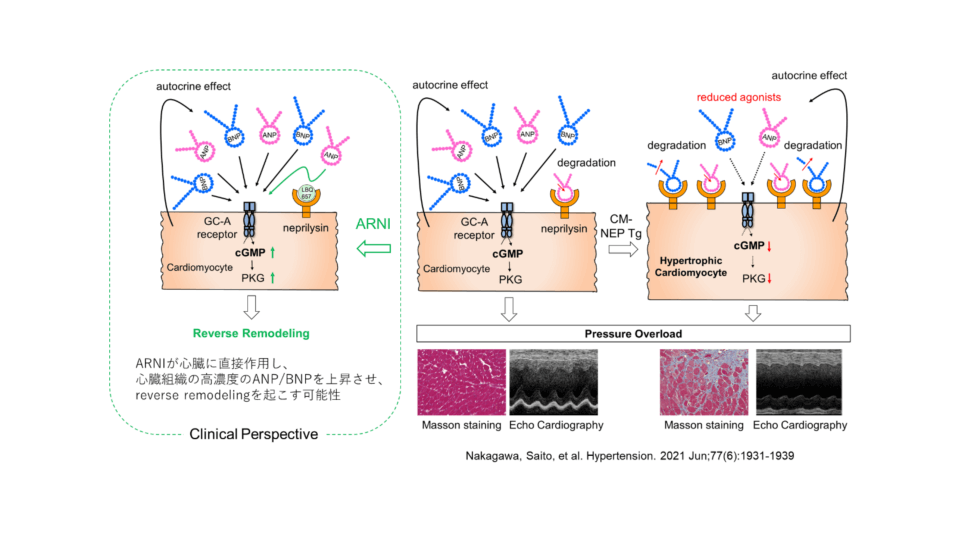
図7 心筋特異的ネプリライシン過剰発現マウス(CM-NEP Tg)の表現型と
clinical perspective.
左:ARNIは直接心臓組織のNEPを阻害することにより、高濃度の組織のANP・BNPの作用を効率的に亢進させることができ、心臓リモデリングを改善することが示唆される。
以上が、最近の主な循環器内科基礎研究の概要およびトピックスです。




